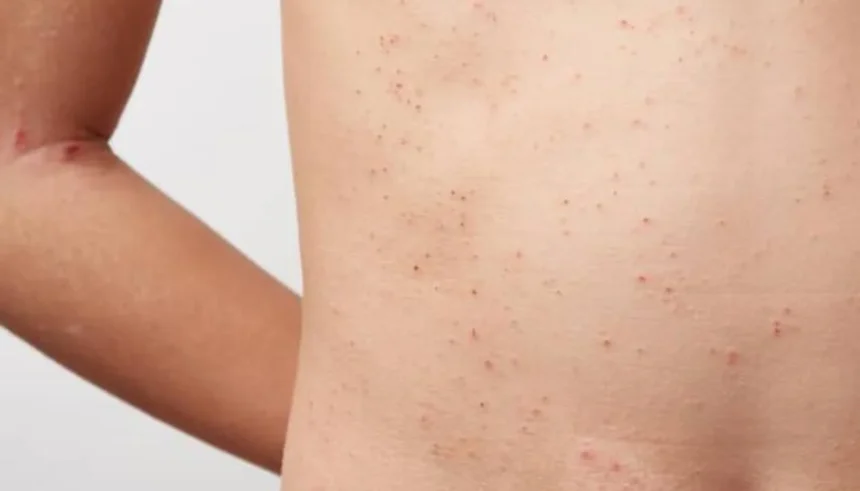
Argentina logró un hito en el campo de la salud al desarrollar el primer biosimilar de la región para el tratamiento de la enfermedad de Fabry. Se trata de una de las terapias de reemplazo enzimático con agalsidasa beta, destinada a abordar esta patología causada por la deficiencia total o parcial de la enzima alfa-galactosidasa A (α-Gal A), cuya función es degradar un lípido denominado globotriaosilceramida (Gb3 o GL3).
“El déficit de esta enzima produce la acumulación de glicolípidos en estructuras intracelulares llamadas lisosomas. Este depósito termina provocando daño en distintos órganos”, explicó el doctor Juan Manuel Politei (MN 102.741), neurólogo y miembro de la Fundación SPINE.
En la misma línea, el médico nefrólogo Fernando Perretta (MN 102.695) advirtió que los pacientes pueden padecer un accidente cerebrovascular a edad temprana, miocardiopatía hipertrófica —conocida como “corazón grande”— o insuficiencia renal que los lleve a requerir diálisis cerca de los 40 años.
La enfermedad de Fabry está catalogada entre las denominadas enfermedades poco frecuentes (EPOF), que son aquellas cuya prevalencia es igual o menor a un caso cada 2.000 nacidos.
“En Argentina se calcula que existen entre 1.200 y 1.500 pacientes, de los cuales unos 900 recibieron diagnóstico y alrededor de 600 están en tratamiento. Si bien los tiempos para llegar al diagnóstico se acortaron en los últimos años, continúa siendo una enfermedad de difícil detección”, detalló Politei.
Perretta agregó que, debido a sus síntomas inespecíficos, la llamada “odisea diagnóstica” puede extenderse incluso durante dos décadas.
Existen dos variantes de esta patología: la forma clásica, que aparece en la infancia, y la forma tardía, que suele manifestarse en la adultez. Detectarla en edades tempranas resulta clave para mitigar las secuelas a largo plazo.
En los niños, el diagnóstico suele darse gracias a que los pediatras identifican signos y síntomas característicos. En cambio, en los adultos, los indicios aparecen entre los 30 y 40 años, cuando la enfermedad ya ha avanzado y afecta órganos vitales como el corazón o los riñones.
Sobre el impacto cardíaco, el cardiólogo Gustavo Cabrera (MP 57.362), responsable del Departamento de Enfermedades Lisosomales del Centro Médico Santa María de la Salud de San Isidro, señaló: “Aunque puede confundirse con otras patologías, hay ‘banderas rojas’ que permiten detectarla. En nuestro ámbito hubo un fuerte trabajo de divulgación científica y se implementaron estrategias como el análisis en pacientes con miocardiopatía hipertrófica o hipertrofia ventricular izquierda sin causa aparente, ya que el 1% de ellos suele tener Fabry”.
La enfermedad afecta tanto a hombres como a mujeres, pero presenta un patrón de herencia ligado al cromosoma X, lo que genera manifestaciones más evidentes en los varones.
“En los hombres, al contar con un único cromosoma X, los síntomas son más notorios. En cambio, en las mujeres, la duplicación de este cromosoma permite que el cuadro pueda ser más leve”, precisó el cardiólogo riojano Juan Muraro (MP 2.046).
Dado su carácter hereditario, los especialistas subrayan la importancia de evaluar a toda la familia cuando se realiza un diagnóstico inicial.
El tratamiento para esta enfermedad es relativamente reciente: recién a partir de la década del 2000 se aprobaron las terapias de reemplazo enzimático (TRE), a las que ahora se suma el desarrollo de este primer biosimilar regional.